A new article from the lab of Prof Tara Spires-Jones, UK DRI at Edinburgh, has shown that, in a mouse model of Alzheimer’s disease (AD), amyloid-beta and tau protein work cooperatively to cause a hyperactivity behavioural phenotype and downregulation of gene transcription relevant to synaptic function. Furthermore, tau reduction corrected these changes suggesting a possible route to therapeutic intervention.
Published today (10 Dec 2019) in Cell Reports, the study aimed to explore the contribution and interaction of the two highly implicated proteins in synaptic dysfunction - an early event in disease pathogenesis and strong correlate to cognitive decline. To overcome previous limitations related to differences in mouse and human tau, and the ability to control tau expression, the researchers utilised a new AD model (MAPTnull+APP/PS1_rTg21221) which overexpresses amyloid-beta, lacks endogenous mouse tau and reversibly expresses human wild-type tau - increasing relevancy to human disease.
The main findings indicated an age-related hyperactivity profile in these mice compared to controls, as measured by open field testing, and significant transcriptional changes, particularly related to increases in neuroinflammation (e.g. complement system) and downregulation of genes in glutamate receptor and calcium signalling pathways suggesting deficits in synaptic function. The authors suggest that a mechanistic link between these changes may involve phagocytosis of synapses by microglia.
To investigate these changes further, Tara and her team suppressed human tau expression using doxycyline (65% reduction), resulting in amelioration of the hyperactivity phenotype and gene expression changes observed previously. This appeared to be a result of preventing further worsening of phenotype rather than recovery. Although tau suppression did not influence associated gliosis in this model, inflammatory markers expressed by glia were reduced including those relevant to the complement system. The team postulate that these changes may indirectly influence the recovery of synaptic gene expression observed. Tau suppression did not change amyloid plaque pathology and soluble amyloid-beta 42 levels, but a reduction in synaptic tau levels was observed, perhaps explaining the improvements in hyperactivity phenotype.
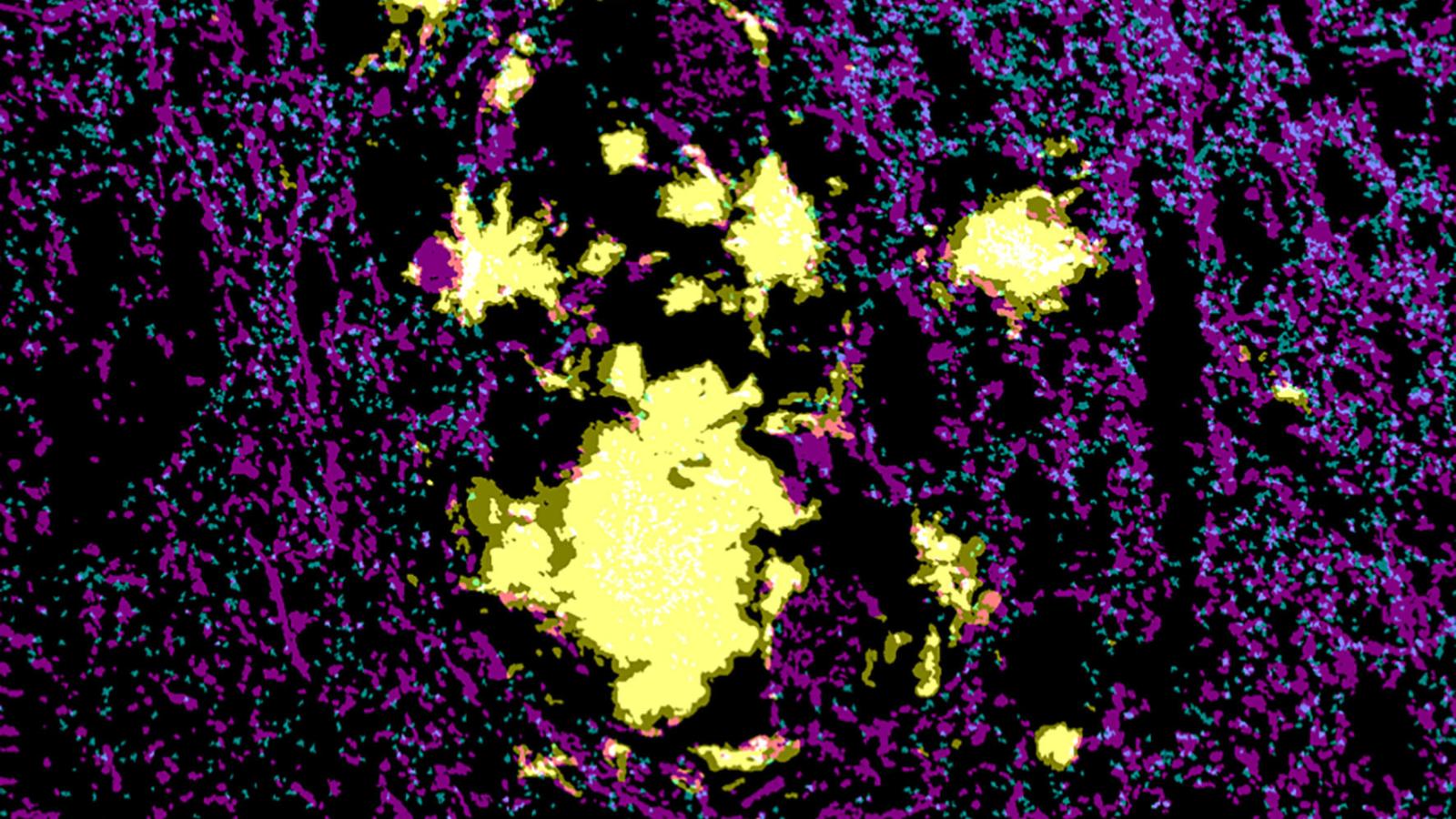
The team additionally investigated human post-mortem brain tissue from people with Alzheimer’s disease to determine translational relevance, showing a greater number of synapses near plaques were positive for the amyloid-beta in contrast to tau localisation which was not influenced by proximity to the plaques. This would suggest similar changes are observed between the model and human disease.
On what the findings mean for our knowledge of Alzheimer’s disease, Prof Tara Spires-Jones said:
"We were intrigued by the the clear neuro-immune signal which was only associated with a behavioural phenotype and synaptic gene transcription reductions when tau was present. This implies that amyloid pathology creates an inflammatory milieu in the brain which is particularly toxic to synapses, with tau playing a significant role in this process."
On plans for future experiments, Tara added:
"We also have a related paper coming out soon which supports the translational relevance of this animal study. In it we observe similar increases in proteins involved in neuron-glia signalling and decreases in proteins important for synaptic function in synaptoneurosomes from human AD brain (bioRxiv, Hesse et al Acta Neuropathologica, in press). We plan to follow up this work in human cells in culture to try and drill into pathways that may prevent or reverse synapse damage."
Reference:
This study was funded by funded by the European Research Council, and the UK Dementia Research Institute which is funded by the UK Medical Research Council, Alzheimer’ Society and Alzheimer’s Research UK.
Article published: 10 December 2019