Researchers led by Prof Sarah Tabrizi (UK DRI at UCL, UCL Queen Square Institute of Neurology and Huntington’s Disease Centre) in collaboration with Prof Gabriel Balmus (UK DRI at Cambridge) and others, have taken major steps forward in advancing genetic therapies for Huntington’s disease. The study, published in the journal Science Translational Medicine, lays an important foundation for future clinical trials targeting the genetic cause of the condition.
What was the challenge?
Huntington’s disease is a devastating neurodegenerative condition affecting movement, thinking and behaviour. It is caused by repetitive expansions of three DNA blocks (C, A and G) in the huntingtin gene. In people who have Huntington’s, this sequence gets longer throughout life until it becomes toxic to cells, leading to the progressive loss of neurons and accelerating neurodegeneration.
There are currently no disease-modifying treatments available for Huntington’s. However, recent studies have highlighted the role of DNA repair in influencing the age of onset and disease progression. MSH3 is a DNA mismatch repair protein, which, in attempting to repair the unusual structures formed by long CAG repeats, inadvertently worsens the disease by further lengthening the CAG repeat tract. MSH3 has been identified as a promising therapeutic target, but more research is needed to determine how low the levels of the protein need to be to suppress CAG repeat expansion.
Targeting MSH3 is exciting not only because it’s directly involved in the CAG repeat expansion, but also because genetic studies suggest that loss of MSH3 function is relatively well-tolerated in humans. Our findings emphasise the potential for MSH3 suppression as a safe and effective way to delay Huntington’s disease.
Group Leader
What did the team do and what did they find?
To determine the level of MSH3 lowering required to have a meaningful impact on CAG repeat expansion, the team used cells taken from a person with Huntington’s, and converted them into neurons in the lab. The neurons were treated with an antisense oligonucleotide (ASO), which binds to MSH3 RNA causing it to be degraded before it can be made into a protein, reducing the levels of MSH3.
The researchers showed that lowering the levels of the MSH3 reduced CAG repeat expansion in the huntingtin gene, in a dose dependent way. Lowering MSH3 by 41% halved the expansion rate and lowering it by 83% was sufficient to completely halt expansion. They demonstrated that lowering MSH3 did not disrupt DNA repair pathways, or trigger cancer signalling pathways.
The team then developed a mouse model with the human MSH3 gene, to enable further testing of the safety and efficacy of MSH3-targeting drugs.
What is the impact?
These results provide valuable information for future clinical trials aimed at developing disease-modifying treatments for Huntington’s disease. The promising strategy could slow or halt Huntington’s at the root of its genetic cause.
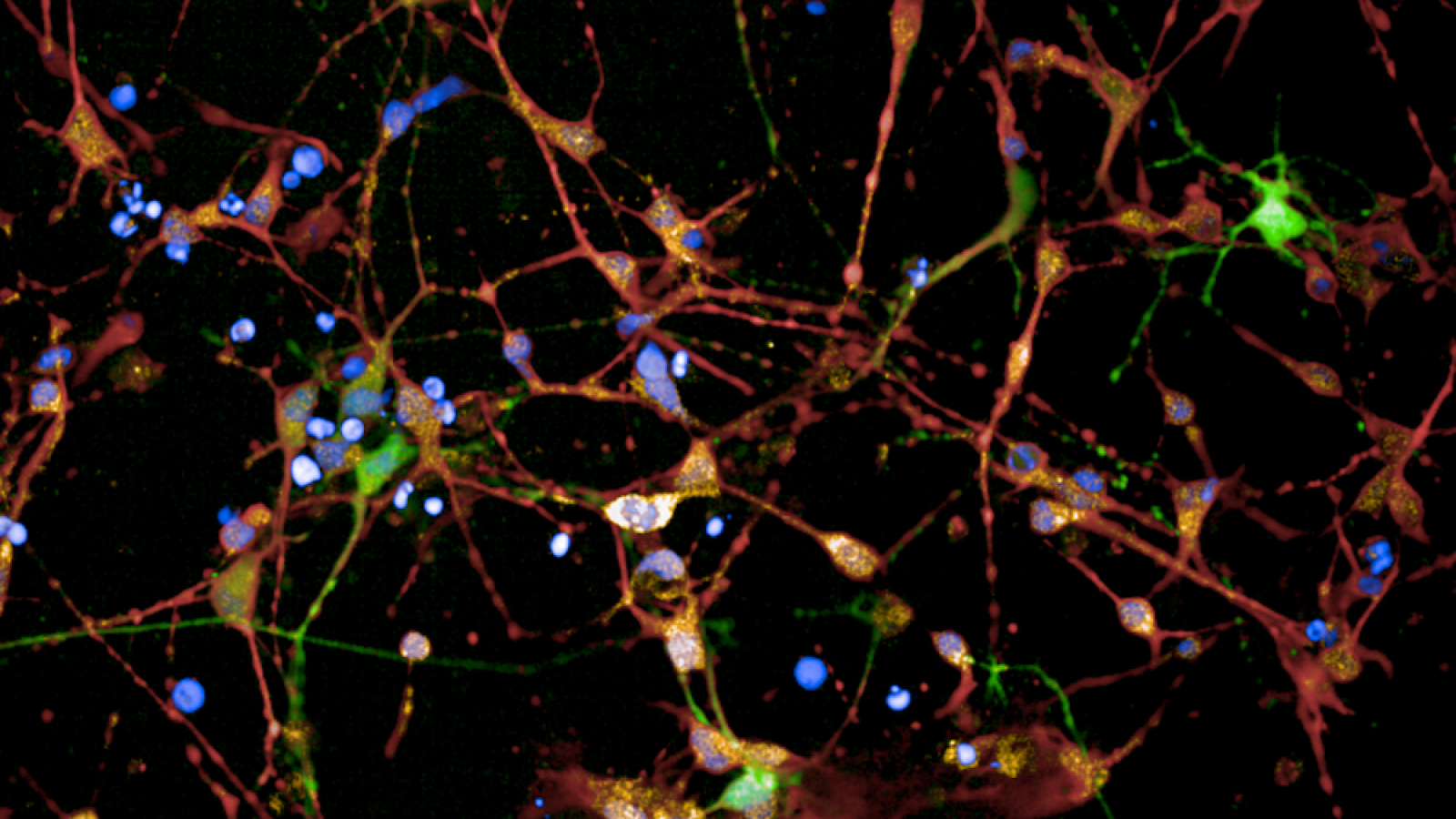
Banner image: Immunofluorescent staining of HD patient 125 CAG iPSC-derived striatal neuron cultures following 36 days of differentiation. Credit: Figure adapted/reproduced with permission from Bunting EL, Donaldson J, et al., Antisense oligonucleotide-mediated MSH3 suppression reduces somatic CAG repeat expansion in Huntington's patient iPSC-derived striatal neurons, Science Translational Medicine, 2025.
Reference: Emma L. Bunting et al., Antisense oligonucleotide–mediated MSH3 suppression reduces somatic CAG repeat expansion in Huntington’s disease iPSC–derived striatal neurons.Sci. Transl. Med.17,eadn4600(2025).DOI:10.1126/scitranslmed.adn4600