The Roger de Spoelberch Prize is awarded for work on neurodegenerative or psychiatric diseases. We are pleased to share that the 2018 prize was awarded to Prof David Rubinsztein, UK DRI at University of Cambridge, for his work on autophagy and neurodegeneration.
Autophagy is a normal physiological process in the body that deals with destruction of cells. The most common neurodegenerative conditions, including Alzheimer’s disease, Parkinson’s disease and Huntington’s disease, are caused by proteins that form aggregates within neurons. The studies from Prof Rubinsztein’s group revealed that autophagy is an important route that cells use to degrade such mutant proteins, including those causing Huntington’s disease (huntingtin), Parkinson’s disease (alpha-synuclein) and forms of dementia (tau).
This team were the first to show that drugs that increase autophagy in cells enhance the removal of mutant proteins causing diseases like Huntington’s disease, spinocerebellar ataxia type 3, Parkinson’s disease and forms of dementia. They have since shown that such drugs are also effective in fruit fly, zebrafish and mouse models of these diseases, in which increased autophagy improves signs of disease. This concept has been subsequently validated in diverse neurodegenerative diseases by many groups. Autophagy is now considered an important neurodegeneration target by pharma.
In order to better understand the biology of mammalian autophagy and identify strategies for therapeutic intervention, Prof Rubinsztein’s team have made detailed cell biological studies of this process and the signals that regulate it. When they began these studies in 2001, rapamycin was the only known drug that could chronically induce autophagy. Although rapamycin is designed for long-term use in humans, it has side effects (unrelated to autophagy) that make it unattractive to patients who may need to take the drug for decades. Therefore, they set out to identify novel autophagy-upregulating compounds by screening drugs previously used in humans for other purposes. This allowed us to identify the first mammalian autophagy control mechanism that is independent of the target of rapamycin. Drugs targeting this were protective in fly and zebrafish models of Huntington’s disease. Rilmenidine, one of the drugs we identified as an autophagy inducer, has beneficial effects in a mouse model of Huntington’s disease and has minimal side effects; this has recently successfully completed a safety trial in Huntington’s disease patients.
Their work has also shown that autophagy compromise contributes to pathology in diverse neurodegenerative diseases, including forms of motor neuron disease, lysosomal storage diseases, polyglutamine expansion diseases like Huntington’s disease, and forms of Parkinson’s disease. Therefore, autophagy deficiency arising from several different primary mutations may be an important contributor to abnormal protein accumulation and cell compromise in these diseases. We will continue to study this important area as part of our UK DRI programme.
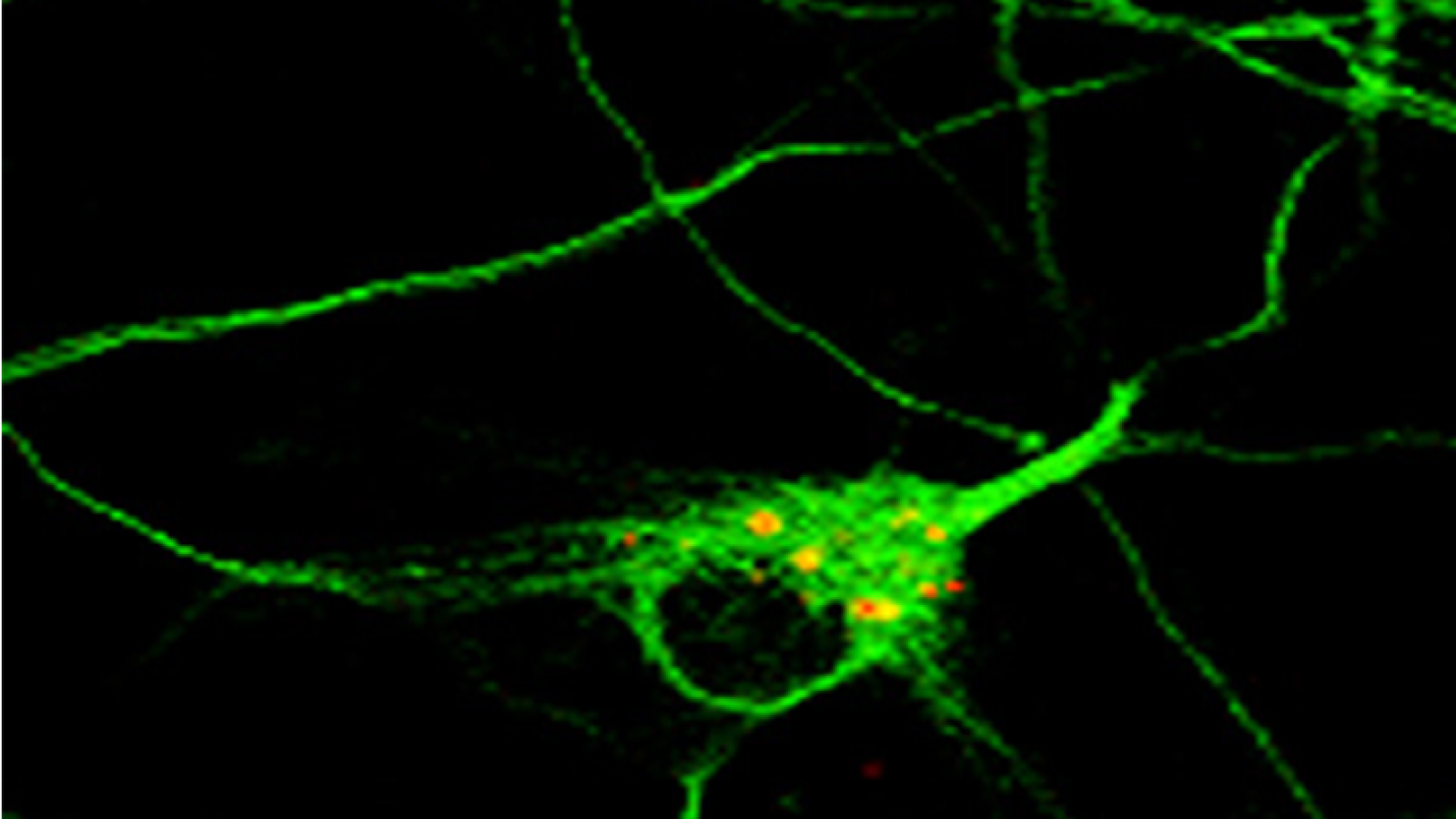
Image caption: autophagosomes in a neuron in primary culture
is awarded for work on autophagy and neurodegeneration
I am extremely grateful to the Roger de Spoelberch Foundation for this great honour. This award will provide tremendous encouragement and significant support for me and my lab’s future efforts. Of course, this prize is a sign of recognition for my coworkers in the lab over the years. I have been truly fortunate to have worked with wonderful people, who are excellent scientists and collaborators. I am very grateful to them for making the lab a place I look forward to coming to each day.Prof David RubinszteinUK DRI at University of Cambridge
