Key details
Repairing DNA to treat Huntington’s disease
Huntington’s Disease (HD) is a genetic disorder that damages nerve cells in the brain due to excessive CAG repeats in the huntingtin gene. This CAG repeat expands over time in brain cells, leading to earlier onset and faster progression of the disease. The Tabrizi Lab research focuses on understanding how DNA repair proteins, which typically fix DNA damage, inadvertently contribute to the expansion of these repeats. By studying these mechanisms, the group aims to find ways to halt this expansion and slow HD progression.
The researchers discovered that the DNA repair protein FAN1 can protect against CAG expansion by interacting with another protein, MLH1. The Tabrizi Lab studies this interaction in detail and test its importance in patient-derived cells using advanced gene editing techniques. Additionally, the team found specific variants of another DNA repair protein, MSH3, which reduce repeat expansion. It will investigate these variants to understand how they alter DNA repair processes.
Furthermore, the researchers are developing innovative methods to measure CAG repeat lengths in cerebrospinal fluid, which could significantly aid clinical trials for new treatments. In collaboration with industry partners, they will screen small molecules and nucleic acid therapies to find potential drugs that modulate DNA repair and prevent repeat expansion. Their goal is to translate these findings into effective treatments for HD and related diseases.
Latest news


Prof Sarah Tabrizi
Prof Sarah Tabrizi FRCP, PhD, FMedSci, FRS is a Group Leader at the UK DRI at UCL. Find out more about her career and expertise on her profile page.

Research summary
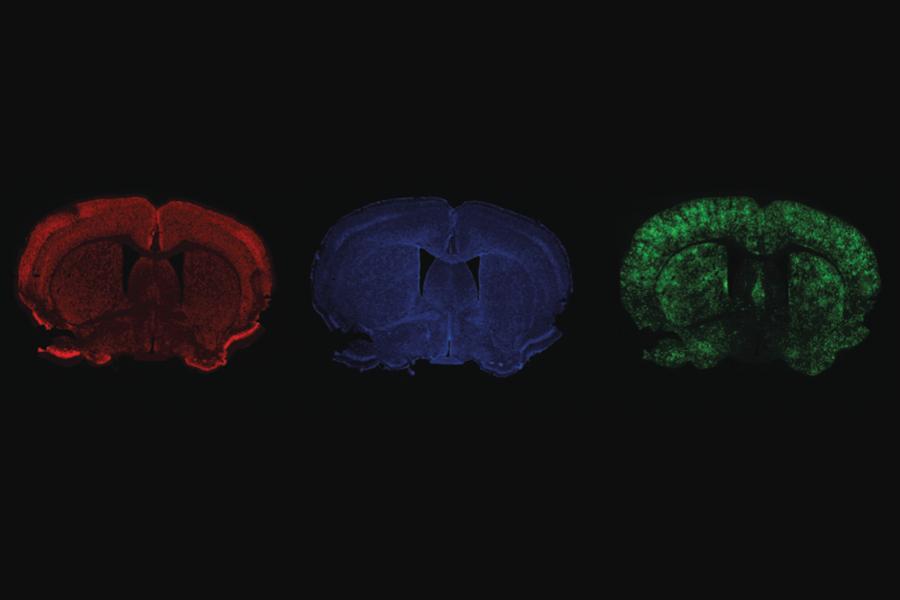
The Tabrizi Lab uses fluorescence to label different cells and structures, e.g. neurons (red) and their nuclei (blue), in the mouse brain.
The mechanism of the DNA damage response in Huntington's disease pathogenesis and relevance for therapeutics
Huntington’s Disease (HD) is an autosomal dominant neurodegenerative disorder caused by an expanded CAG repeat in the HTT gene. This repeat continues to expand throughout life, particularly in vulnerable tissues, leading to earlier onset and more rapid disease progression. The Tabrizi Lab research investigates two fundamental questions: the mechanisms by which DNA repair processes drive CAG repeat expansion and how these processes can be modulated to treat HD and other repeat expansion diseases.
In previous studies, the group demonstrated that FAN1, a DNA repair protein, mitigates CAG repeat expansion in cultured human cells and HD patient medium spiny neurons (MSNs) by competing with MSH3 for MLH1 binding and through its nuclease activity. The researchers are now focused on characterising the interaction between FAN1 and MLH1 using in-cell protein interaction assays and mass spectrometry (MS). They are validating the importance of this interaction in patient-derived induced pluripotent stem cells (iPSCs) with CRISPR-introduced mutations in the interaction and nuclease domains. Additionally, they are investigating the role of FAN1’s post-translational modifications (PTMs) in regulating its interactions and repeat stabilisation activity. The team is also assessing the role of FAN1 in other repeat expansion diseases, such as Spinocerebellar Ataxia type 3 (SCA3), C9orf72-related amyotrophic lateral sclerosis/frontotemporal dementia (ALS/FTD), and Fragile X Syndrome (FXS), by knocking down FAN1 in iPSCs derived from patients with these conditions.
The Tabrizi Lab identified MSH3 genetic variants that reduce repeat expansion and slow HD progression. It is modelling these variants, as well as mutations in the ATPase and MLH1 binding domains, in U2OS cells and HD MSNs and studying their impact on DNA and protein interactions, and on repeat instability. Genetic knockout and ASO-mediated knockdown of MSH3 effectively halted CAG expansion and may induce repeat contraction. As loss of MSH3 is well-tolerated, unlike MSH2 and MSH6, the group is developing novel frameshift reporter assays to specifically monitor MSH3 activity.
The researchers have shown CAG expansion is slowed by reducing HTT transcription through the repeat. This was achieved by mutation of the promoter and introduction of bidirectional transcriptional repressors in a U2OS cell model. They are now assessing the role of transcription in HD patient iPSC-derived medium spiny neurons (MSNs) by CRISPRi-mediated knockdown and promoter disruption and Levels R-loops, which form at repeats during transcription, increased with repeat length in these MSNs.
To address the clinical need for biomarkers of CAG instability, the team is developing ultrasensitive assays to size the HTT CAG repeat from cerebrospinal fluid (CSF), validating them in patient samples.
The researchers' comprehensive approach aims to elucidate the underlying mechanisms of HD pathogenesis and translate these findings into effective therapies, in collaboration with numerous academic and industry partners.
Key publications
Vacancies
Lab members

Collaborators






Lab funders
Thank you to all those who support the Tabrizi Lab!
