Researchers led by Prof Giampietro Schiavo, UK DRI at UCL, have used two new mouse models of motor neurone disease (MND) to show that early disease mechanisms may differ depending on the causative gene. Deficits in transport along nerve cells preceded symptoms in one but not the other model, suggesting that therapeutics may have to be tailored to individuals according to their genes.
MND, also known as amyotrophic lateral sclerosis (ALS), is a rare but fatal, progressive neurodegenerative disease resulting in loss of motor neurons, muscle wasting, and ultimately death - most often due to respiratory failure. The disease is thought to develop from a complex interplay between genetic and environmental factors. Mutations in several key genes are known to cause MND that result in faulty versions of proteins essential for life.
A number of disease mechanisms have been investigated in the search for effective treatments, and disruption to the transport network in neurons is an emerging central theme. In this study, published today in Cell Reports, Prof Schiavo, Dr James Sleigh and their teams set out to explore this phenomenon in two new transgenic mouse models of MND caused by faulty TDP-43 or FUS proteins. TDP-43 and FUS have several similarities and common functions in healthy brain, suggesting that they may share disease mechanisms.
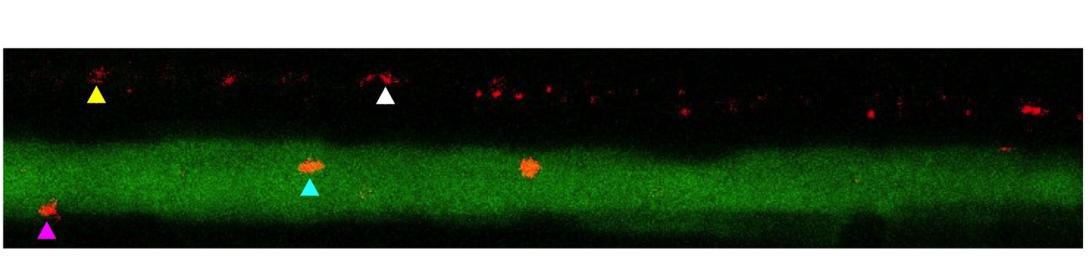
Researchers can track the movement of important survival signals around the cell
In order for neurons to remain healthy and survive, they must constantly traffic and deliver key cargoes up and down their long, thin processes. Signalling endosomes are one such cargo, which transport survival signals from the distant regions of the nerve cells to their centres.
Using a sophisticated new method performed in mice, the group were able to track these signalling endosomes within motor neurons in real time, to determine whether any problems were occurring as the disease developed. In the TDP-43 mice, which show MND-related pathology without loss of motor neurons, they found deficits in signalling endosome trafficking before motor symptoms, suggesting its importance as an early disease mechanism. In contrast, the FUS mice showed few issues with transport along nerves, despite displaying motor neuron loss.
The results imply that, although transport deficits in the motor neuron may contribute to disease symptoms, this is not universal to all mutations and genes implicated in MND, for example FUS. With the caveat that these experiments were performed in animal models, which only mimic some aspects of human disease, the study provides greater understanding of the mechanisms behind MND and perhaps fundamental differences between individuals affected.
On the study, Prof Giampietro Schiavo, said,
“This study is important because it demonstrates that several mechanisms contribute to MND pathology, and that therefore we should implement targeted, mechanism-oriented approaches to developing personalised therapies for this devastating disease.”
Lead author, Dr James Sleigh, added,
“The significance of transport to nerve cell survival is indisputable; however, to determine its relevance to the early stages of devastating neurological diseases like ALS we need to study this dynamic process in appropriate research models. Our study is important because it highlights a key disparity in contributing mechanisms between two major ALS causing genes, while emphasising the critical nature of studying disease mechanisms on a whole-organism scale.”
Reference:
Article published: 17 March 2020