A new paper from the lab of Prof John Hardy, UK DRI at UCL, in collaboration with Prof Valentina Escott-Price, UK DRI at Cardiff, has identified four new risk genes for Alzheimer’s disease, tied to the microglial response to amyloid beta. We sat down with lead author, Dr Dervis Salih, to find out more about the study published in Brain Communications and how it contributes to our understanding of dementia.

with Dr Dervis Salih
(Postdoctoral Researcher in the lab of Prof John Hardy, UK DRI at UCL)
Hi Dervis, can you give us some background on this study?
The last decade of Genome Wide Association Studies (GWAS) has revealed the initially surprising results that many of the risk genes for late-onset Alzheimer’s disease (AD) are expressed mostly by microglia, and not neurons. Additionally, twin studies suggest that 60-70% of risk for AD is hereditary, but the 30+ risk genes that have been identified using GWAS do not account for all of the genetic risk. Therefore, we wanted to investigate whether genetic variability in the microglial response to amyloid deposition influences risk of AD. It is critical that we find better approaches to analyse the rich data we have from GWAS and understand how these loci are connected in genetic pathways and biological function.
What were your main findings?
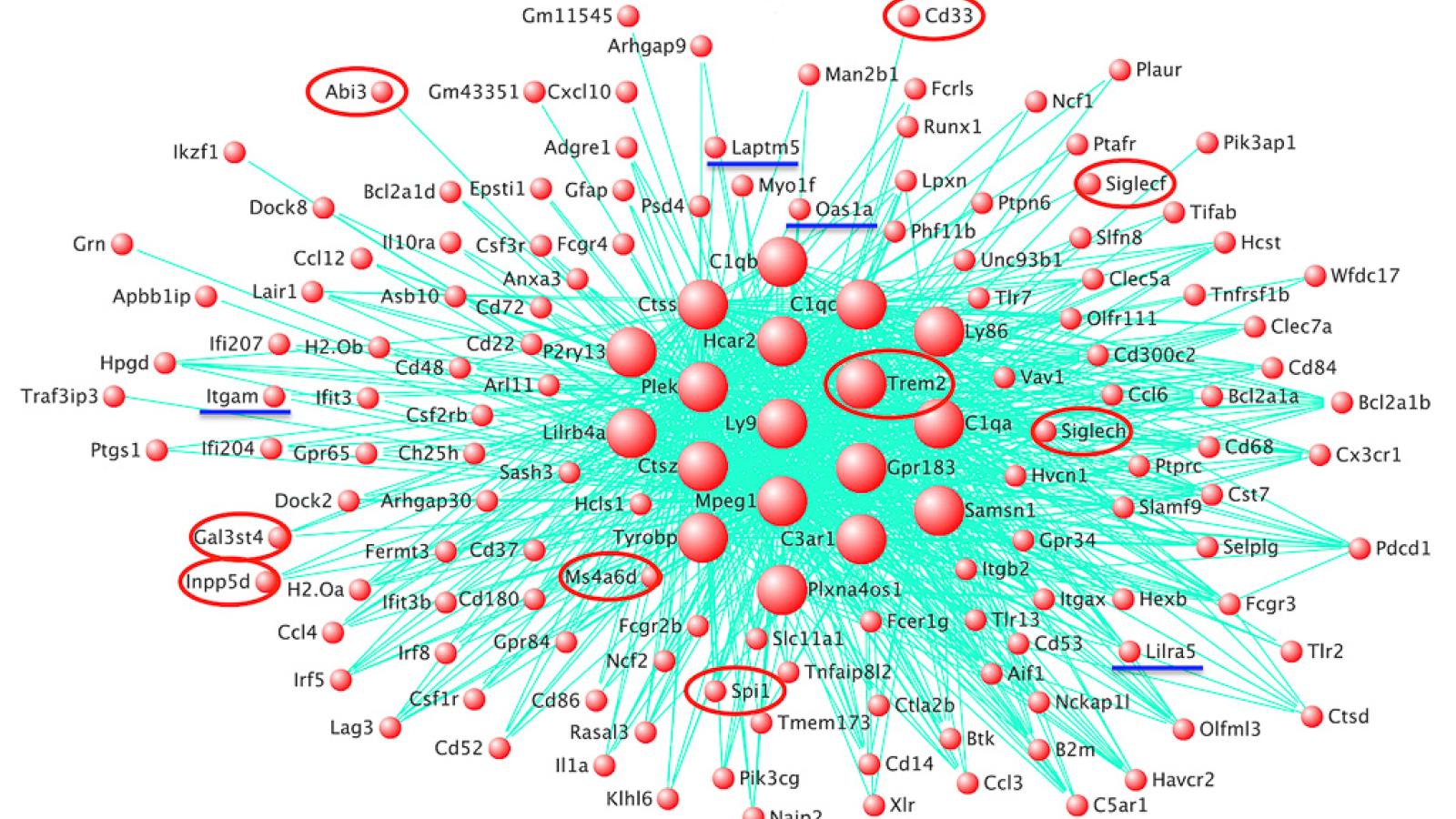
Our findings supported the idea that variability in genes expressed by microglia significantly influence the risk of developing AD. Using our new approach, we identified at least four putative risk genes for AD (OAS1, LAPTM5, LILRB4 and ITGAM) at a high statistical stringency.
We saw that orthologues for the established risk genes for AD, including TREM2, ABI3, PLCG2 and SPI1, and our new putative risk genes, formed a co-expression network in amyloid-responsive microglia in transgenic mice. There was a significant enrichment of genes with variation in the human population within this network.
We also saw that the microglia responding to tau pathology engaged a different co-expression network, providing evidence that amyloid and tau pathology stimulate microglia in different ways.
What method did you employ to investigate this?
We identified the four candidate risk genes by combining two methods. Firstly, we rescored the GWAS single nucleotide polymorphism (SNP) data (Kunkle et al. (2019)) in a method developed by Prof Valentina Escott-Price at UK DRI at Cardiff.
We then performed RNA-seq in transgenic mice with amyloid pathology to assess the expression of most genes in the hippocampus, and used a co-expression network analysis algorithm to identify modules of gene-expression associated with amyloid deposition in mice. A network of genes expressed by microglia correlated most strongly to amyloid deposition.
Finally, we statistically compared the genes showing significant ‘gene-level’ variation from the rescored GWAS study with the gene orthologues from the co-expression network to give us the four candidate risk genes, and verified the importance of several established risk genes such as TREM2 and SPI1.
The critical advance of this paper was combining the rescored human GWAS data with the RNA-seq analysis in transgenic mice. Traditional GWAS methods overlook more complex relationships between DNA variation and disease.
Are there any patterns to the networks of risk genes you identified?
Yes, the co-expression network is driven at least in part by the core microglial transcription factor SPI1, shown recently to contain a common SNP in the human population that protects against AD. Our data also suggests the existence of auxiliary, environment-dependent transcription factors that modify disease development, but further work is required to identify these.
We did not see an overwhelming effect of pro-inflammatory activation in the transgenic mice associated with pathology at the transcriptome level. However, it is tempting to speculate that ITGAM/CD11b, LAPTM5 and LILRB4 are involved in phagocytic processes by microglia related to synaptic re-modelling during disease progression.
Interesting, the identification of OAS1 and the presence of the other interferon-related genes within the network of amyloid-responsive microglia suggests a relatively unexplored role for interferon signalling in AD, perhaps involved in limiting viral infections, recruiting immune cells to sites of damage and/or regulating cytokine production.
Recognising that gene expression in mouse and human microglia differs, what’s the implication for your study and any other mouse-model studies?
We see a significant enrichment of gene-level variation from humans associated with disease in the gene orthologues expressed by mouse microglia, even when established risk genes for AD are removed. This means a significant part of the microglial response to amyloid is conserved between humans and mice.
We also compared the genes expressed abundantly in human microglia, using two independent datasets, with the genes present in our amyloid-responsive microglial co-expression network in mice, observing substantial overlap.
However, there are some important differences and moving forward the correct model needs to be selected for the experiment or genes in question.
Finally, what questions do you and the field still need to address and what are your plans for future work?
Aside from improved model organisms and in vitro models of human AD, we need to identify more of the risk genes associated with disease by improving the methods and models we have used here. We need to confirm, with high-throughput sequencing, how the candidate genes we identified vary in people with disease i.e. is the association due to more than a simple SNP?
We also need to understand how risk genes are connected in one or more microglial pathways, and some of this may be resolved by studying chromatin organisation and epigenetic changes in microglia in health and disease (Novikova et al. BioRχiv (2019)).
Producing a more complete model of human AD in mouse models (amyloid-dependent tauopathy, without mutations to tau), will likely only be possible by combining a number of manipulations, genetic and environmental. For example, on top of the amyloid deposition, we will probably need to induce blood-brain barrier breakdown, prime the immune system by exposure to environmental pathogens, alter diet, alter APOE status, attenuate Trem2 signalling or even humanise several genes.
Our future work will be to study the molecular roles of our putative risk genes in microglia. By understanding these microglial pathways in more detail, we will be better equipped for drug discovery to slow disease progression, even allowing us to ascertain disease risk at a personal level. It is likely that different AD patients will have different subtypes of disease, and will require tailored treatments.