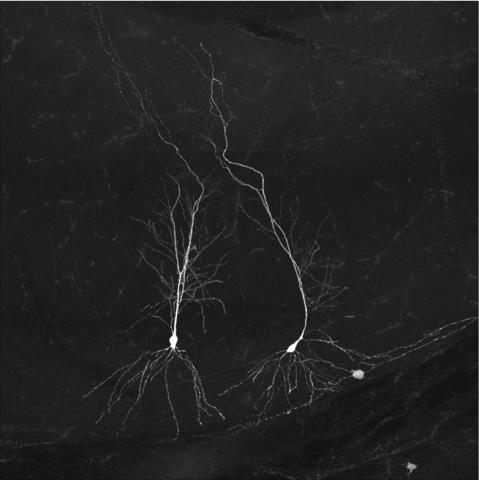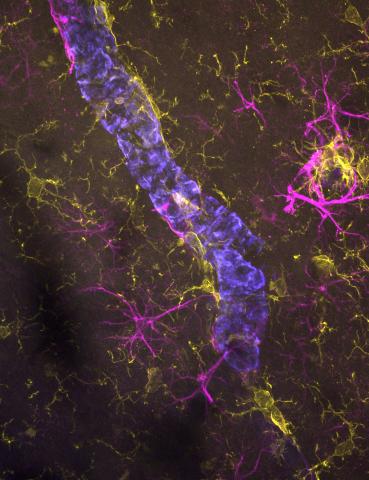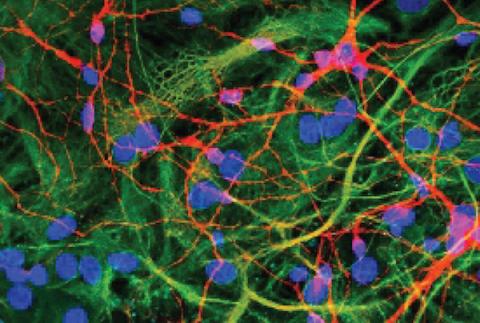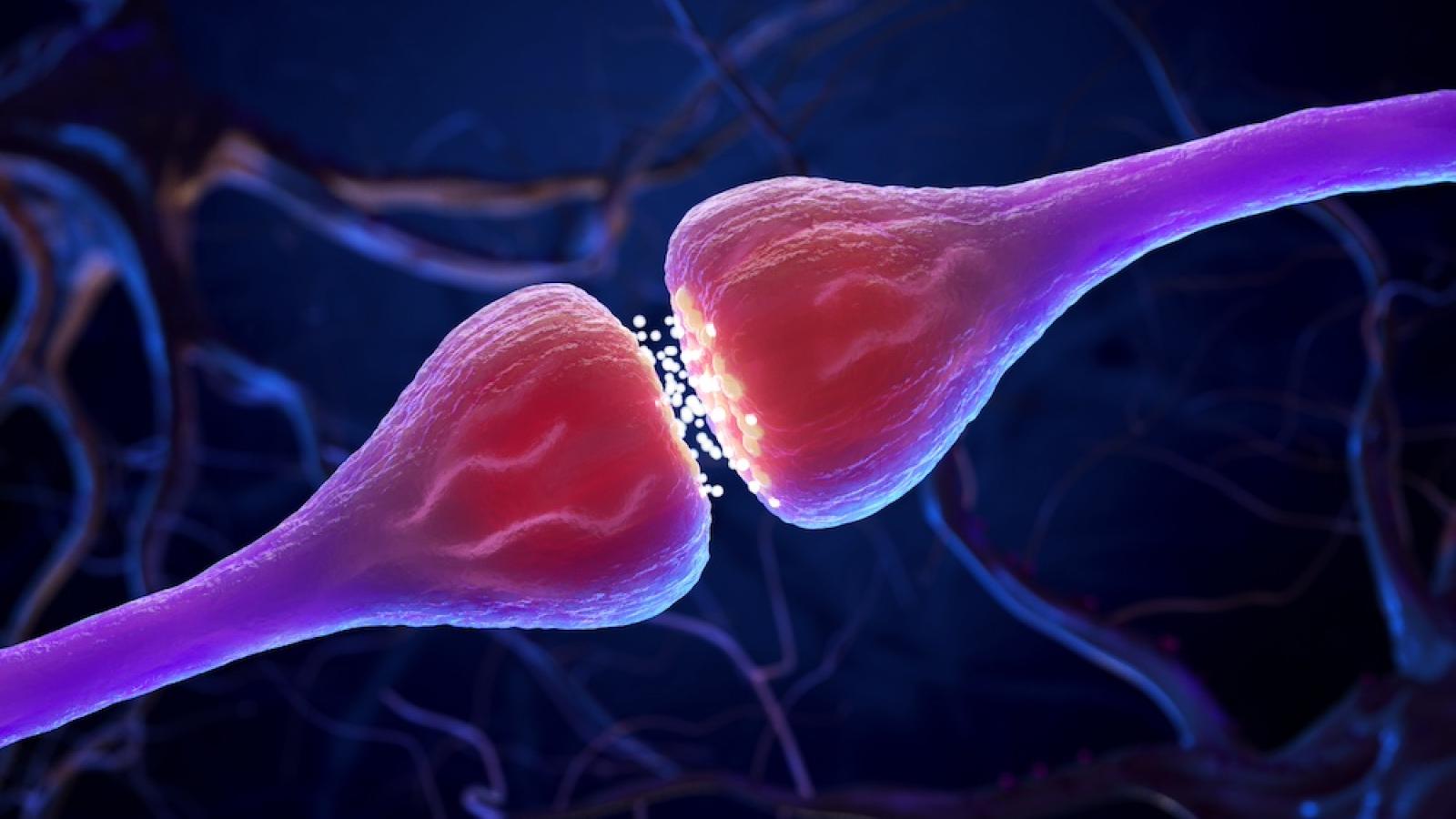
The remarkable computational power of the brain is partly due to its unique structure, where billions of individual brain cells connect up to form circuits via tiny contact points known as synapses. Synapses facilitate the flow of electro-chemical signals in a coordinated manner and are an essential part of the nervous system and its optimal functioning. Synapses within the central nervous system are integral for supporting memory, learning and perception while those in the peripheral nervous system play an important role in executing muscle movement through specialised structure known as the neuromuscular junction.
Synapse dysfunction and loss are the hallmark pathophysiology of all neurodegenerative conditions. However, synapses are highly diverse structures and as a result are selectively vulnerable across different neurodegenerative conditions. For example, Alzheimer’s disease is characterised by synapse loss in regions associated with learning and memory, while in motor neuron disease, neuromuscular junctions are lost causing issues such as muscle weakness.
It is clear that better understanding of why synapses are selectively lost across neurodegenerative disease could unlock effective new treatments for a range of conditions. Researchers at the UK DRI are unpicking the molecular cascade leading to synapse dysfunction and loss, from the single structure through to wider neural network level, with the ultimate aim of making discoveries that could drive the next generation of therapeutics.
Latest news

A deeper dive
Synapse dysfunction and loss are fundamental to the pathophysiology of neurodegenerative conditions and correlate strongly with poor cognitive outcome. For example, some of the earliest signs of Alzheimer’s disease are changes to electrical activity patterns within regions of the brain that are key for learning and memory. Although the molecular causes behind this abnormal brain activity are not yet understood, they may contribute to the gradual loss of synapses between neurons. Our understanding of the processes that mediate these changes in electrical activity, how it leads to synapse dysfunction and elimination, and the repair and compensatory mechanisms within the neuron to counter and compensate for the synapse loss remain largely unexplored.
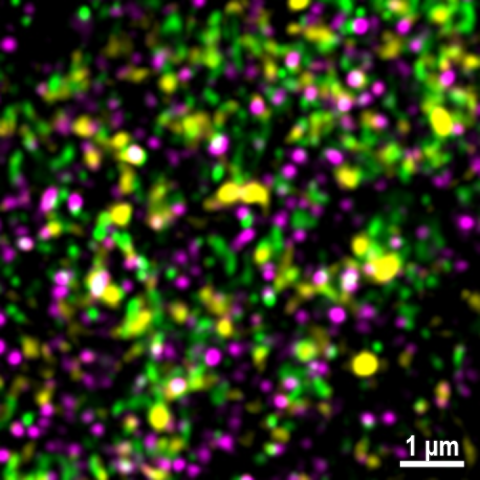
Investigation into synapse biology is difficult as they exist in highly complex structures composed of thousands of distinct proteins, which makes characterisation challenging. Synapse types have traditionally been discriminated based on neurotransmitter molecules, but it is now clear that protein composition exhibits deeper diversity within traditional neurotransmitter types. Such diversity is further complicated by additional factors, such as brain-state dependent changes in synaptic activity, regional susceptibility to disease pathology and interplay with non-neuronal cell-types.
At the UK DRI we are interested in uncovering the mechanisms underlying synaptic loss and dysregulation of network activity. In particular we are interested in:
- molecular composition of synapses in health and disease states (Grant, Jackson, Matthews)
- the role of toxic aggregates on synapse function (Klenerman, Bowles, Barnes, Avezov, Durrant, Connor-Robson, Beccano-Kelly)
- the processes that mediate synapse loss, including the role of the microglia in this process (Hong, Morgan, Spires-Jones, Zelek)
- the transsynaptic spread of toxic aggregates (Spires-Jones, Duff, Schiavo, Klenerman)
- the effects of the toxic aggregates at a network (Barnes, Opazo, Busche, Gan, Beccano-Kelly) and behavioural (Wiseman, Krupic) level.
- brain-state dependent changes in synaptic activity (Dijk, Busche, Wisden, Brancaccio, Grossman, Jaramillo)
We are also exploring opportunities to translate our findings through non-invasive neuromodulatory interventions (Grossman) and the development of biomarkers that measure synapse degeneration (Sogorb-Esteve).