What is Huntington's disease?
Huntington’s disease (also referred to as HD) is a condition that affects the body’s central nervous system, causing damage to nerve cells in the brain, which gets worse over time. This damage affects a person's movements, thinking ability and behaviour.
It’s a genetic condition, which means those who have Huntington’s disease have inherited it from one or (on rare occasions) both parents. Symptoms usually develop between the ages of 30 and 50 although they can start earlier or later in life. Huntington’s disease affects both men and women.
UK DRI Group Leader Prof Sarah Tabrizi discusses Huntington's disease and her work on pioneering treatments for the condition. In 2023, the pharmaceutical company Roche began a phase II Generation HD2 trial of the drug tominersen in people at the earlier stages of disease, when it is thought to be most effective.
Frequently asked questions
- How common is Huntington's disease?
Huntington’s disease is caused by a faulty gene that is passed on from parent to child, so people are usually only at risk if one or both parents carry the gene. 1 in 10,000 people in the UK is affected by Huntington’s disease1.
Every child conceived naturally to a parent with the gene has a 50% chance of inheriting it2. This also means they have a 50% chance of not inheriting the gene, in which case they would not develop the condition and would not be able to pass the gene on to any children they may have.
Although it is possible to develop Huntington’s disease without a known family history of the disease this is most likely to be because it wasn’t diagnosed in the parent.
- What are the symptoms of Huntington's disease?
Symptoms can vary greatly between individuals, even within the same family, but they cover three main areas:
- Movement: This can include involuntary movements and having less control over what you want to do.
- Cognition: Such as difficulty thinking or concentrating and memory lapses.
- Behaviour: Changes in personality or mood swings. These can be the most distressing symptoms for the person living with Huntington’s disease and their loved ones.
Early indications that the condition could be present are clumsiness, lack of concentration, difficulty remembering things, and mood swings. As the condition progresses other symptoms can include:
- Involuntary jerking and/or fidgety movements of the limbs and body
- Increasing difficulties with speech
- Problems with balance, walking and co-ordination, leading to increasing difficulties with mobility
- Problems with swallowing and difficulties maintaining weight
- Further changes to thinking and behaviour such as impulsivity, apathy and lack of insight
- Increasing dependence on others for activities of daily living
During the later stages of the disease full time nursing care is necessary.
More information about what to look out for can be found on the Huntington’s Disease Association and NHS websites.
- How is Huntington's disease diagnosed?
If you’re concerned that you or someone you care about may be showing Huntington’s disease symptoms, the first step is to make an appointment to see a GP. They may refer you or your loved one to a specialist for tests which could include a brain scan. The specialist will discuss the symptoms and rule out other neurological and neurodegenerative conditions. Ultimately, a genetic test from a blood sample will confirm a Huntington’s disease diagnosis; this is known as diagnostic testing.
If there is a family history of the condition then it is possible to have a genetic test (after the age of 18) in the absence of any symptoms to see whether the faulty gene has been inherited. This is known as presymptomatic testing. It is a very personal choice and many people choose not to take the test and instead wait to see whether symptoms arise. Anyone who does consider taking the test is encouraged to have at least three sessions of genetic counselling before taking the test, during which they will have chance to discuss the implications before making a final decision.- What treatments are available for Huntington's?
As of summer 2024, there are no approved treatments that specifically target Huntington’s disease. However, there are options available to reduce the symptoms caused by the disease. These include:
- Medication: For example, there are medications which can alleviate symptoms of involuntary movements, and antidepressants may be prescribed to treat depression. It’s important to note that people respond differently to medications and what is helpful for one person may not be for another.
- Aids to support daily life: An occupational therapist can recommend equipment that may make it easier to live with Huntington’s disease, such as mobility aids, ramps, grab rails and voice activated products.
- Help with eating and communicating: A speech and language therapist and dietician can work with people living with Huntington’s disease to develop bespoke solutions to address problems with speech, swallowing and weight loss.
- Physiotherapy: This can help with mobility issues as staying safely active for as long as possible contributes to better physical and mental health.
For people affected by Huntington’s disease, information on getting involved in research including clinical trials can be found on the UCL and Huntington’s Disease Association websites.
- Support for Huntington's disease
Support is crucial, not only for those living with Huntington’s disease, but also for their families and carers. The Huntington’s Disease Association and Genetic Alliance websites have useful information and links to resources about regional support, the process of genetic testing, starting a family, and other aspects of living with Huntington’s disease. The Brain Charity also offers counselling to support those with a diagnosis.
Further information about support is available on our ‘support for people affected by dementia’ page.
What causes Huntington's disease?
As a genetic condition, Huntington’s disease is caused by a mutation in an individual’s DNA. In 1993, scientists identified the specific gene, ‘huntingtin’, and a mutation was found which causes a particular coding sequence known as CAG to repeat. This repeat triggers a faulty, toxic form of the huntingtin protein to accumulate, which damages neurons in the brain, eventually leading to neurodegeneration and clinical symptoms.
While there is currently no cure for Huntington’s disease, science is advancing all the time and researchers from the UK DRI are working hard on new discoveries to help treat the condition and slow symptoms.
UK DRI Group Leader and clinical neuroscientist Prof Sarah Tabrizi is at the forefront of developing therapies for the Huntington’s community including tominersen, an ‘antisense oligonucleotide’ drug, that aims to target and reduce harmful huntingtin protein. The phase II Generation HD2 trial is testing the treatment in people at the earlier stages of disease, when it is thought to be most effective. In September 2025, it was announced that patients receiving the a new gene therapy treatment experienced 75% less progression of the disease overall, compared to a matched cohort who did not receive the treatment. This was the first time a drug trial has reported continuing, statistically significant slowing of Huntington’s progression. The global trial was sponsored by uniQure and the trial team included Prof Tabrizi.
Prof Vincent Dion is harnessing CRISPR technology to edit genes directly and has shown in exciting proof-of-concept studies that he is able to ‘contract’ the expanded CAG repeat region in Huntington’s which could slow the progression of Huntington’s disease.
Another promising area of research for Huntington’s disease relates to mechanisms of DNA repair which are thought to go wrong in the condition. Prof Gabriel Balmus is leading efforts to identify drug targets to increase DNA repair that could be utilised in future treatments.
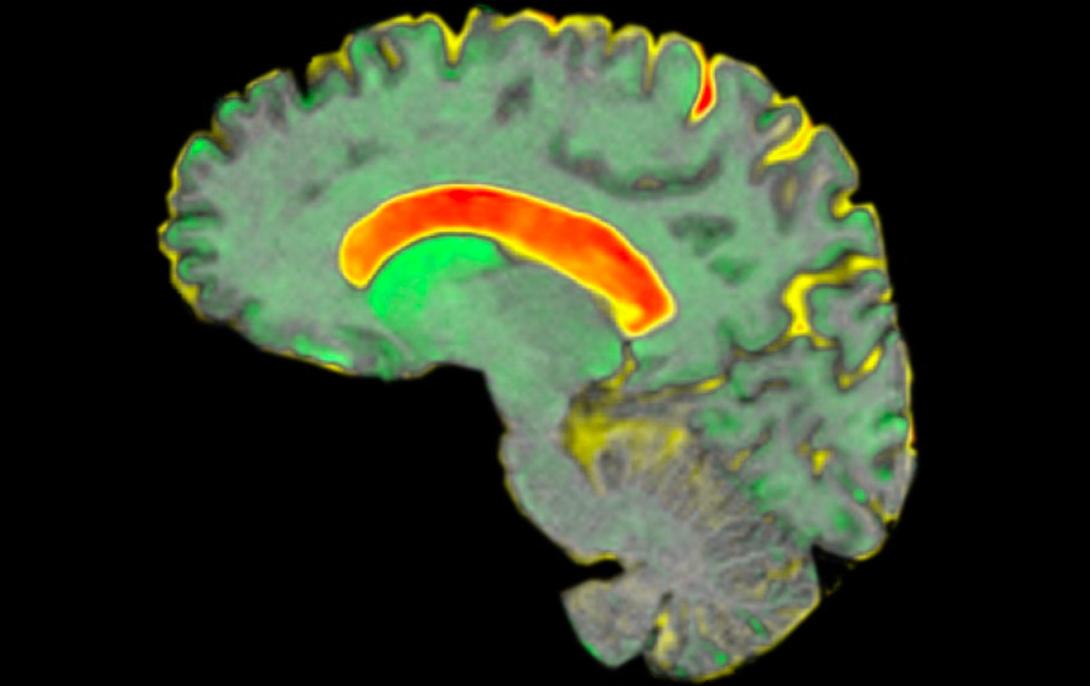
A brain. scan from the Track-HD study. Credit: Dr Rachael Scahill
Latest news
Labs working on Huntington's