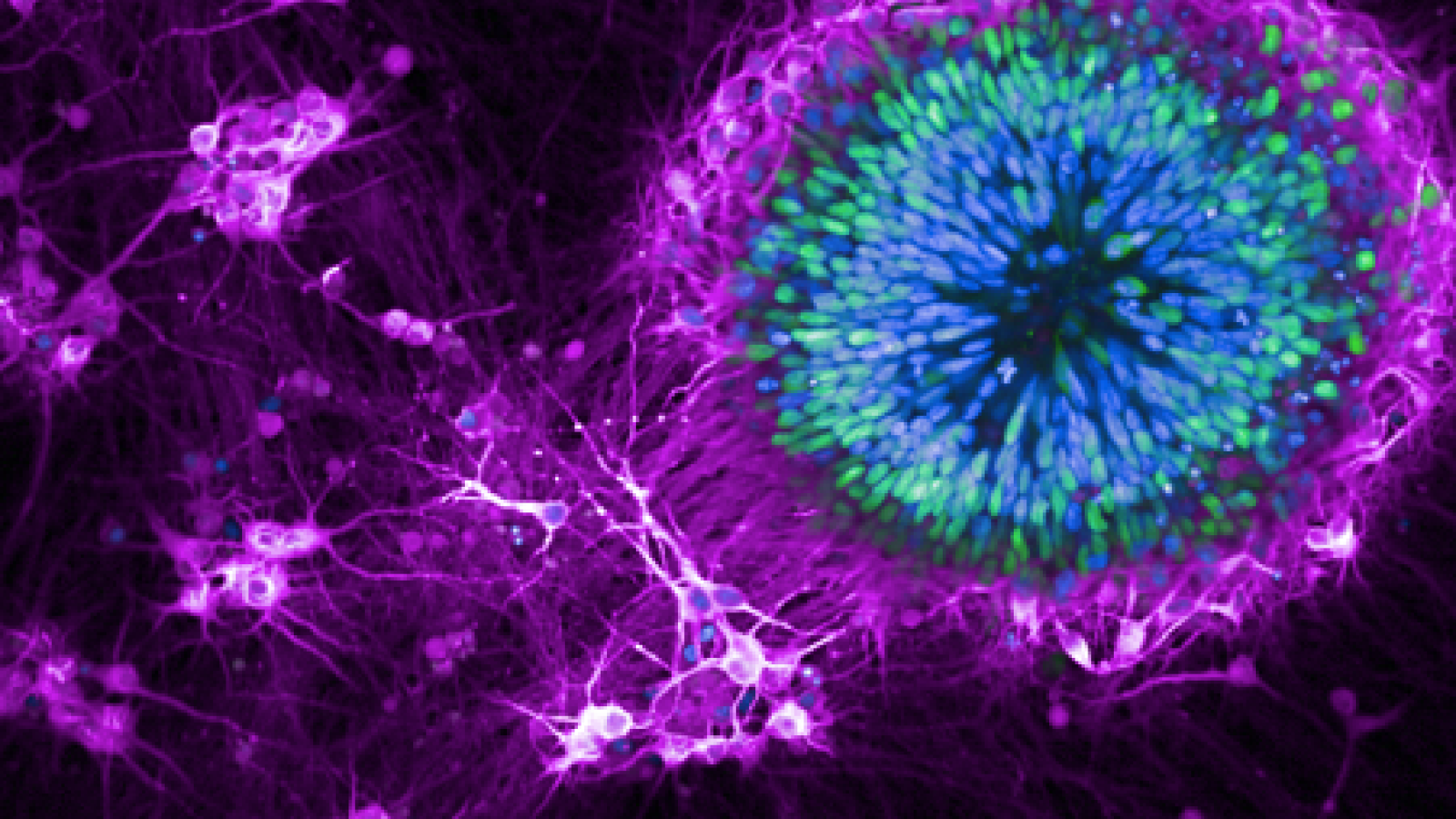
A key role for neuronal transport disruption in disease
Frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS, the most common form of motor neurone disease (MND)) are devastating and progressive neurodegenerative diseases. Common symptoms to arise in FTD include behavioural and personality changes, and in ALS include issues with movement and breathing. However, increasing evidence suggests that they actually share many features, perhaps including their molecular cause.
Faults in the C9orf72 gene are the most common genetic cause of both conditions and are found in around 1 in 10 patients with FTD and the same proportion of people with ALS. Genes code for proteins in the body and therefore a fault in these instructions can lead to a dysfunctional and potentially harmful protein. The C9orf72 fault also produces new repetitive proteins which are highly toxic.
Scientists do not yet fully understand how these repetitive proteins cause the death of neurons, but they may affect transportation to and from the nucleus – the cell’s control centre. If this passage is somehow altered, this may cause the abnormal build-up of protein in the wrong part of the cell forming toxic aggregates – which are linked with neuron damage in both FTD and ALS.
The Mizielinska Lab are delving into the inner-workings of the neuronal transport machinery in exquisite detail. They hope to discover more about what effect C9orf72 fault has on transport within the cells and therefore what may be going wrong in FTD and ALS. Success in these investigations may reveal key targets for the design of new treatments for people with these conditions.
Dr Sarah Mizielinska
Dr Sarah Mizielinska is a Group Leader at the UK DRI at King's. Find out more about her career and expertise on her profile page.

Latest news


Research summary
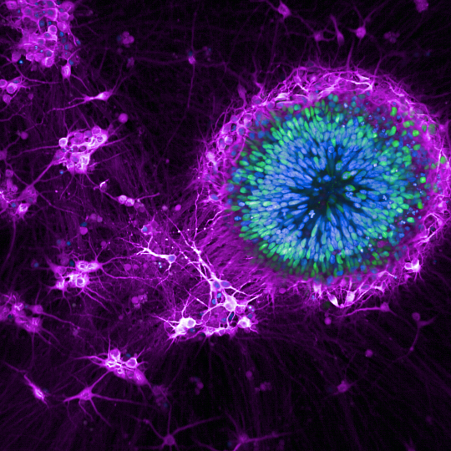
iPSC cells used in MND/FTD research.
Credit: Mizielinska Lab
Single molecule nucleocytoplasmic transport dynamics in FTD/ALS
Frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS) are two clinically different neurodegenerative diseases that lie on the same pathogenic spectrum. The two diseases can be found within the same family, but also co-occur within individuals, and share overlapping genetic causes and pathologies. The most common genetic cause of both diseases is a repeat expansion mutation in the gene C9orf72, which is responsible for around 10% of all FTD and ALS cases and is the most common of all neurodegenerative disease-causing mutations.
The C9orf72 mutation could initiate disease due to toxic novel RNA and protein species emanating from the repeat expansion or by the loss of C9orf72 protein. In vitro and in vivo models show that two of the proteins produced from the repeat – repeating polypeptides of glycine-arginine or proline-arginine – are highly neurotoxic. However, it remains unclear exactly how these proteins exert their neurotoxicity. These repetitive polypeptides are disordered in structure, and therefore, perhaps unsurprisingly, interact with other intrinsically disordered proteins. The disordered nature of these proteins allows them to form multivalent interactions and undergo liquid-liquid phase separation and phase transitions, which forms the basis for cellular compartmentalisation and membraneless organelles. Indeed, in vitro studies show that C9orf72 polypeptides associate with membraneless organelles such as the nucleolus and stress granules and disrupt their function.
This is by no means the only implication of proteins containing intrinsically disordered domains in FTD and ALS. Many proteins that either harbour disease-causing mutations or form pathological aggregates contain intrinsically disordered domains, such as TDP-43, FUS, and hnRNPs, and the mutations often affect the phase separation properties of these proteins. Pathological aggregation of these proteins is a hallmark of FTD/ALS (and other dementias) and is the result of dysfunctional nucleocytoplasmic transport and their consequent mislocalisation from the nucleus to the cytoplasm. Cytoplasmic protein aggregates can also sequester elements of the nucleocytoplasmic transport pathway. This suggests that nucleocytoplasmic transport may be an initiator and propagator of neurodegenerative disease. However, the precise mechanisms by which nucleocytoplasmic transport is affected by the C9orf72 mutation is not yet known.
Main objectives and research goals:
This UK DRI programme led by Sarah Mizielinska will investigate the connection between the dynamics and protein interactions during nucleocytoplasmic transport through the nuclear pore, disease-associated proteins containing low complexity domains (LCDs), and common downstream pathologies:
- How is single-molecule transport through the nuclear pore affected by disease-associated proteins? Using super-resolution microscopy to image single molecules in intact cells to monitor nucleocytoplasmic trafficking dynamics.
- How are the biophysical properties of nuclear pore selection barriers affected in FTD and ALS? Experimental approaches include the use of hydrogel-mimics prepared from phase transitioning of human nuclear pore proteins.
- How is disrupted nucleocytoplasmic transport linked to common disease pathologies and cell death? Multiplexed longitudinal live cell imaging combining nuclear import experiments with other assays.
Key publications
Vacancies
Lab members


Collaborators








Lab funders
Thank you to all those who support the Mizielinska Lab!